PROSPER (PROtease Specificity Prediction servER) is an integrated feature-based web server that provides prediction of novel substrates and their cleavage sites of 24 different protease families from primary sequences. PROSPER addresses the “substrate identification” problem to support understanding of protease biology and development of therapeutics targeting specific protease regulated pathways.
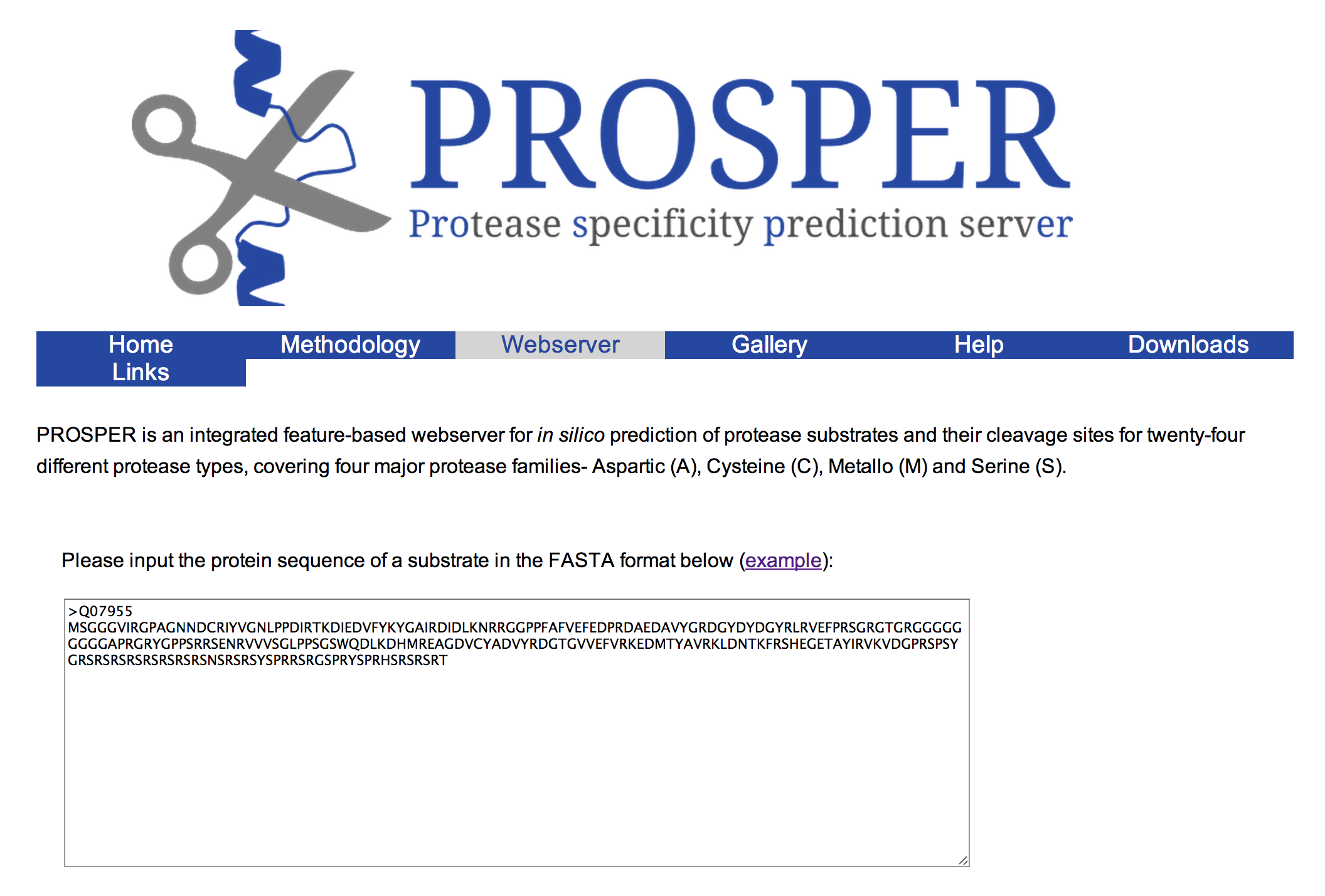
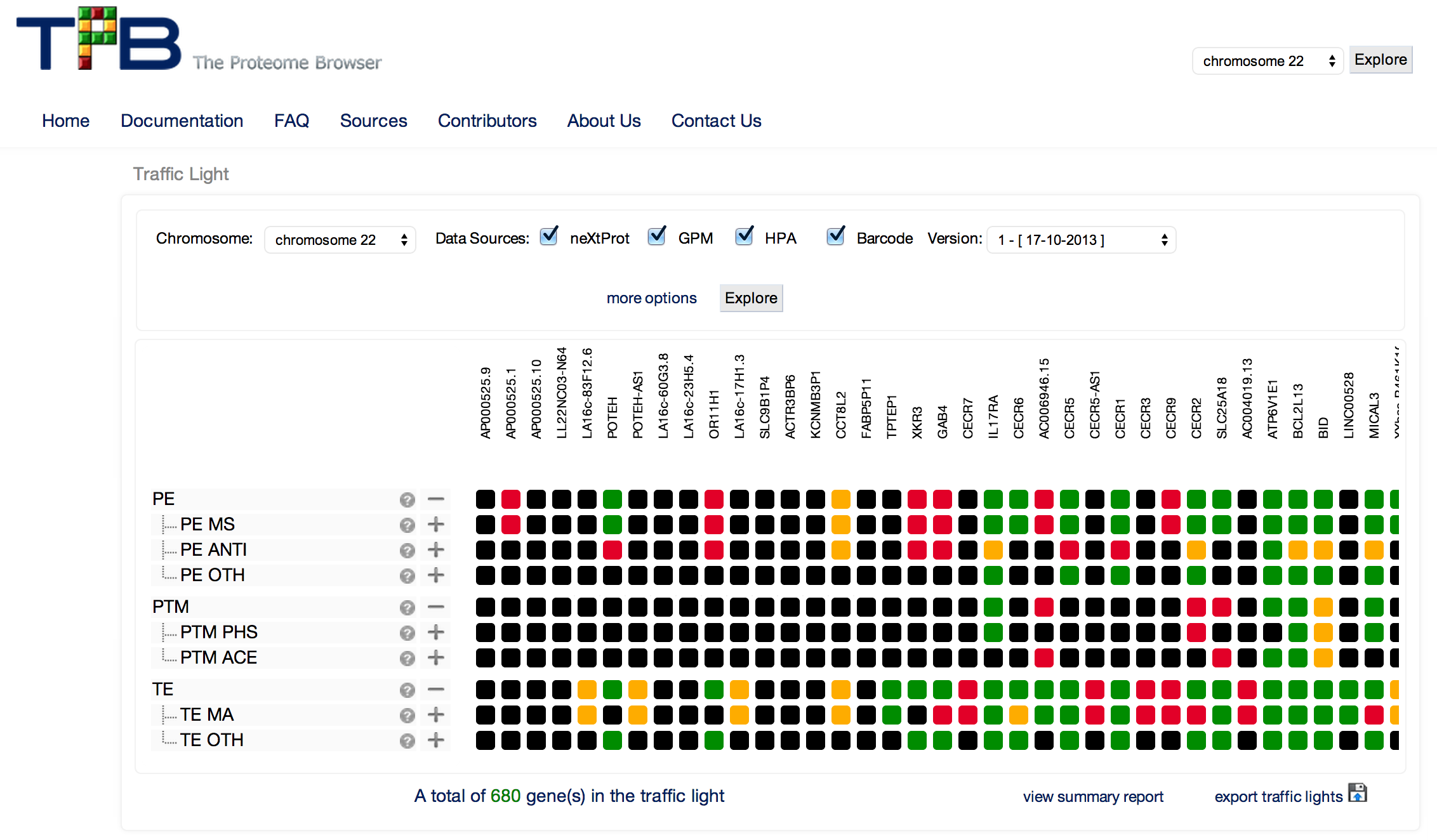
PROSPER’s web server for protein sequence user input.
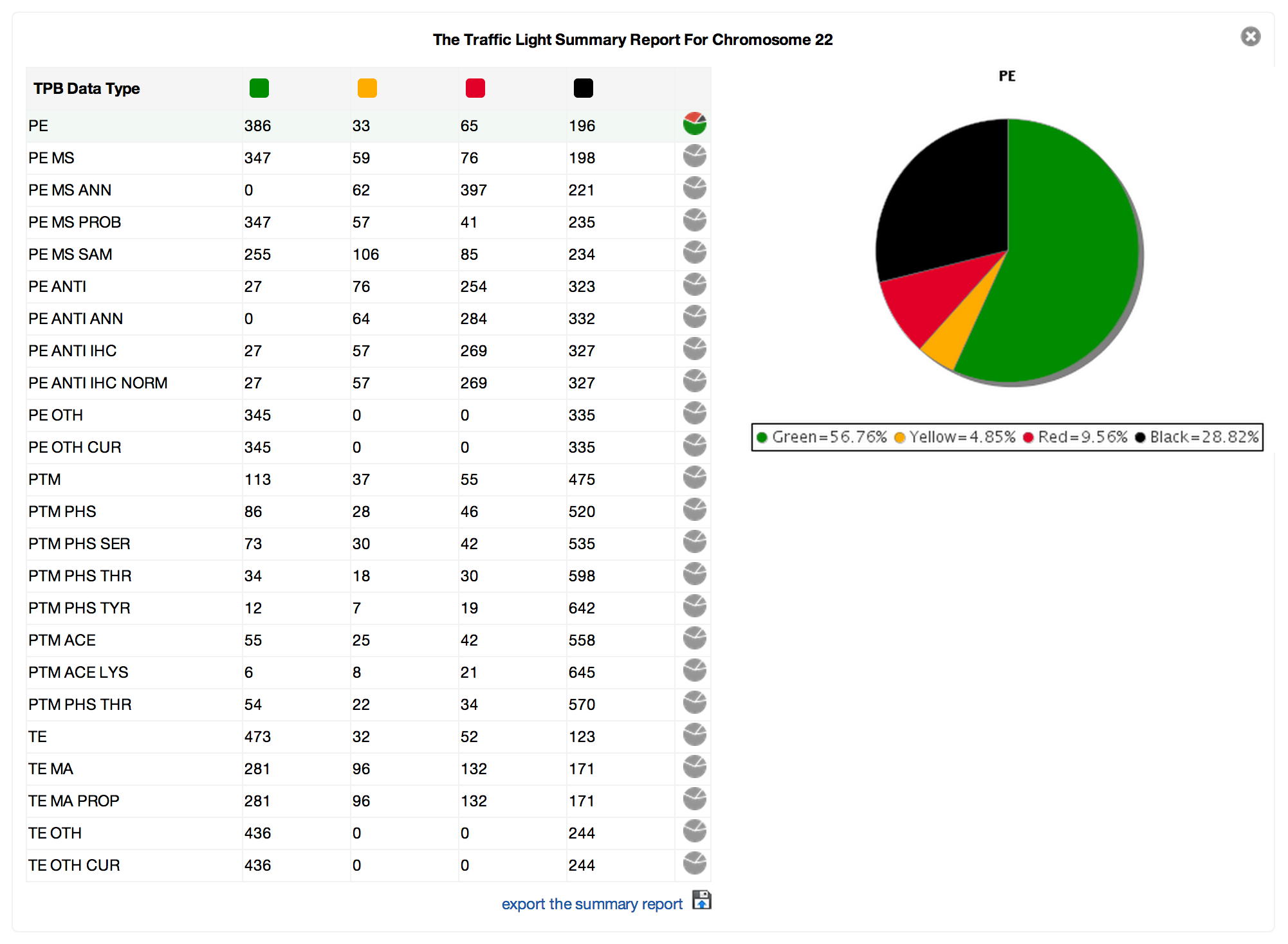
Query sequences in FASTA format are submitted to PROSPER using a simple web interface. PROSPER then uses a machine learning approach based on “support vector regression” to produce real-valued prediction of substrate cleave probability. After the prediction tasks have been performed, PROSPER provides users with a link to access their query sequence’s prediction results.

PROSPER’s result page showing colour-coded predicted cleavage sites.
The R@CMon team helped the Monash Bioinformatics Platform in migrating the PROSPER web server into the NeCTAR Research Cloud. PROSPER is now using persistent storage (Volumes) granted via VicNode computational storage allocation for its model database which currently contains 24 proteases. To date, PROSPER has served more than 6000 users from 68 countries for their own research and these numbers are expected to grow in the near future.
DOUBLE MUTANT PBP2X T338A/M339F FROM STREPTOCOCCUS PNEUMONIAE STRAIN 2 R6 AT 2.4 A RESOLUTION
Future plans for PROSPER include tighter integration with high-performance computing (HPC) facilities and the NeCTAR Research Cloud to enable simultaneous sequence prediction analyses; increase the coverage of the model database from 24 to 50 proteases to support the wider protease biology community; and implementation of an online computational database of PROSPER-predicted novel substrates and cleavage sites in the whole human proteome. The later will facilitate the functional annotation of the complete human proteome and complement with ongoing efforts to characterise their functions.