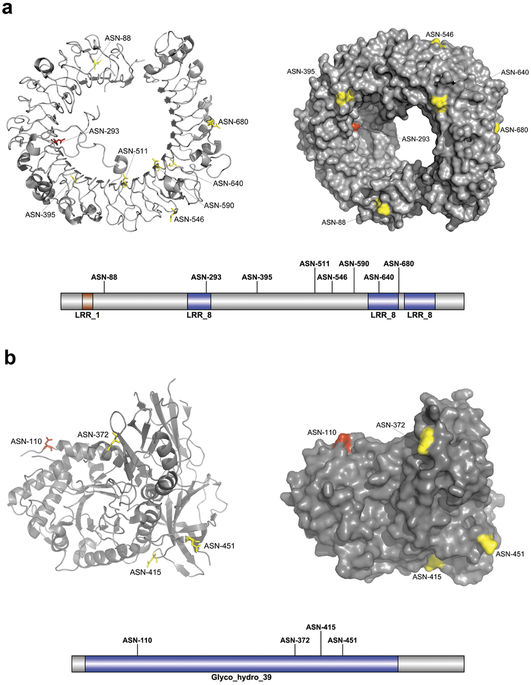
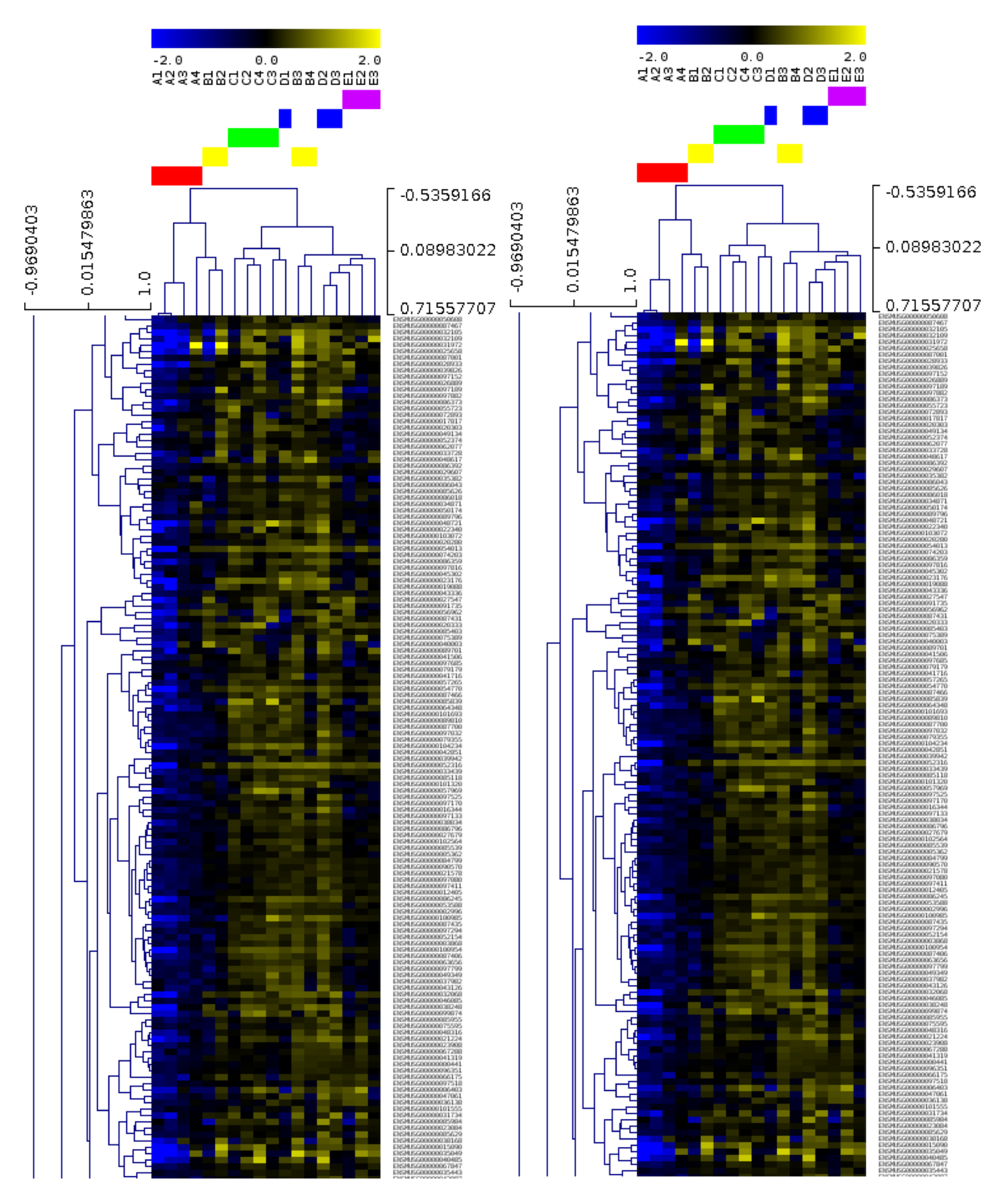
An integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data : the impact of making machine learning good practice readily available to the community.
Associate Professor Jiangning Song is a long-standing user of the Monash Research Cloud (R@CMon). He is the lead of the Song Lab within the Monash Biomedicine Discovery Institute. Jiangning’s journey began with the deployment of the Protease Specificity Prediction Server – PROSPER app in 2014. Since then the lab has launched more than 30 bioinformatics web services, all of which are made available to research communities worldwide.
Their latest contribution, iLearn, addresses key obstacles to the adoption of machine learning applied to sequencing data. Well-annotated DNA, RNA and protein sequence data is increasingly accessible to all biological researchers. However, at the scale of this data it is challenging if not impossible for an individual to manually investigate. Similarly, another obstacle to broad scale access is that investigation and validation through wet laboratory experiments is time consuming and expensive. Hence when presented appropriately, machine learning can play an import role making higher-level biological data accessible to many researchers in the biosciences.
Many of the previous works and tools only focus on a specific step within a data-processing pipeline. The user is then responsible for chaining these tools together, which in most cases is challenging due to incompatibilities between tools and data formats. iLearn has been designed to address these limitations, using common patterns informed by the lab and its collaborators.
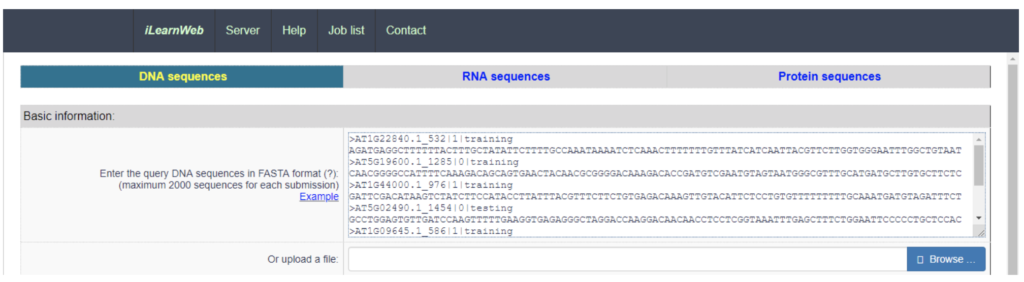
An emerging breakdown of the pipeline steps is:
- Feature extraction
- Clustering
- Normalization
- Selection
- Dimensionality reduction
- Predictor extraction
- Performance evaluation
- Ensemble training
- Results visualisation
iLearn packages these steps for use in two ways. Users can use iLearn through an online environment (web server) or as a stand-alone python toolkit. Whether your interest is in DNA, RNA or protein analysis, iLearn provides a common workflow pattern for all three cases. Users input their sequence data (normally in FASTA format), and then enters various descriptors and parameters for the analysis.
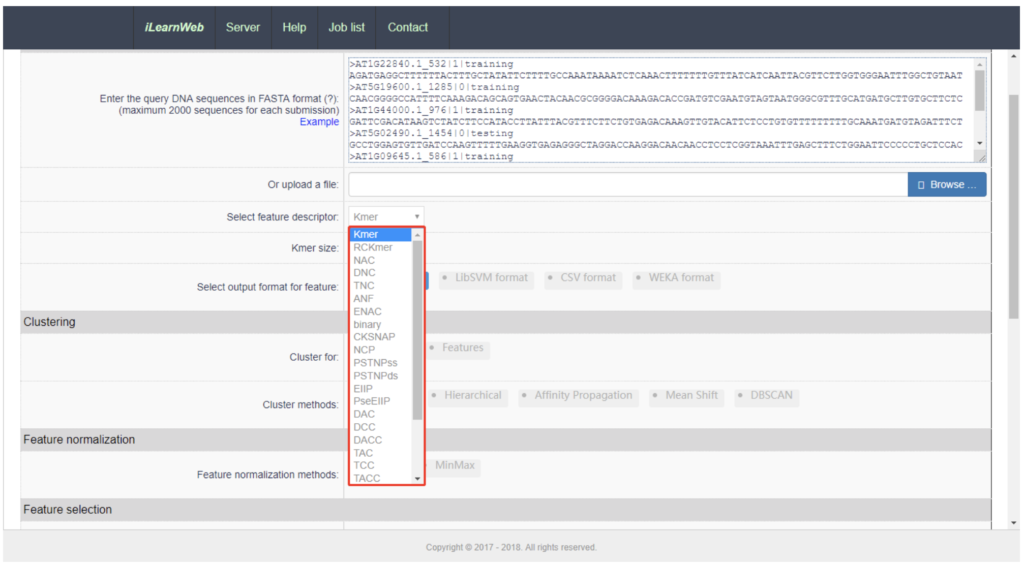
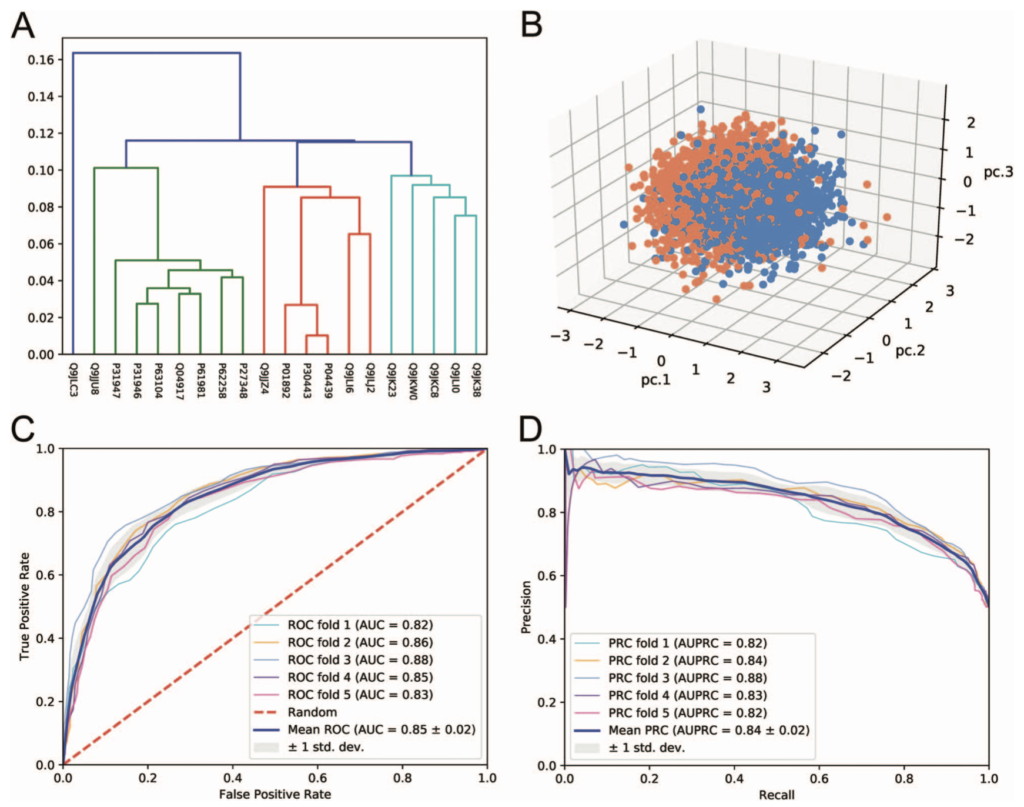
The results page shows the various output, once again informed by the Lab’s good-practices. They can be downloaded from the web server in various formats (e.g CSV, TSV). High quality diagrams and visualisations are also generated by iLearn within the web server:
Since iLearn’s release, more than 5K unique users have used the web server worldwide. The user community and resultant impact continues to grow, with 60 citations since the tool’s seminal publication.
iLearn has been used as an efficient and powerful complementary tool for orchestrating machine-learning-based modelling which in turn improves the speed in biomedical discoveries through genomics and data analysis. As new descriptors get developed and optimised, iLearn aims to incorporate these into future releases to further improve its performance with the R@CMon team providing support to tackle the potential increase in computational and storage complexities.
This article can also be found, published created commons here 1.