The Epigenetics and Chromatin (EpiC) Lab at Monash University is working on understanding how mutations in certain chromatin factors promote the formation of brain tumours. This project involves the generation and analysis of high-throughput sequencing data of chromatin modifications and remodellers in normal and mutated cells. The sequencing is carried out at the MHTP Medical Genomics Facility and the resulting datasets are then imported into the analysis workflow running on the Monash node (R@CMon) of the NeCTAR Research Cloud. The sequencing reads are first aligned to the repetitive fraction of the genome using a script developed by Day et al. (Genome Biology 2010) to determine enrichment at repeats. Sequencing reads are then aligned to the genome using Bowtie. The resulting files are filtered for quality, poor matches and PCR duplicates using customised Perl scripts. The filtered files are then imported into SeqMonk for further analysis.
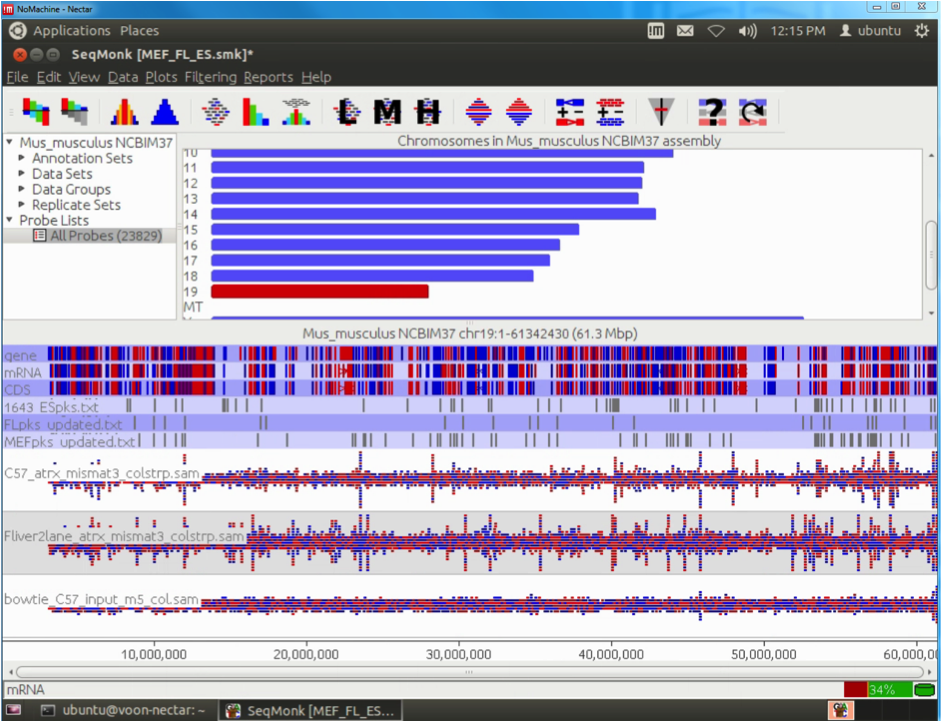
Overlap analysis using SeqMonk
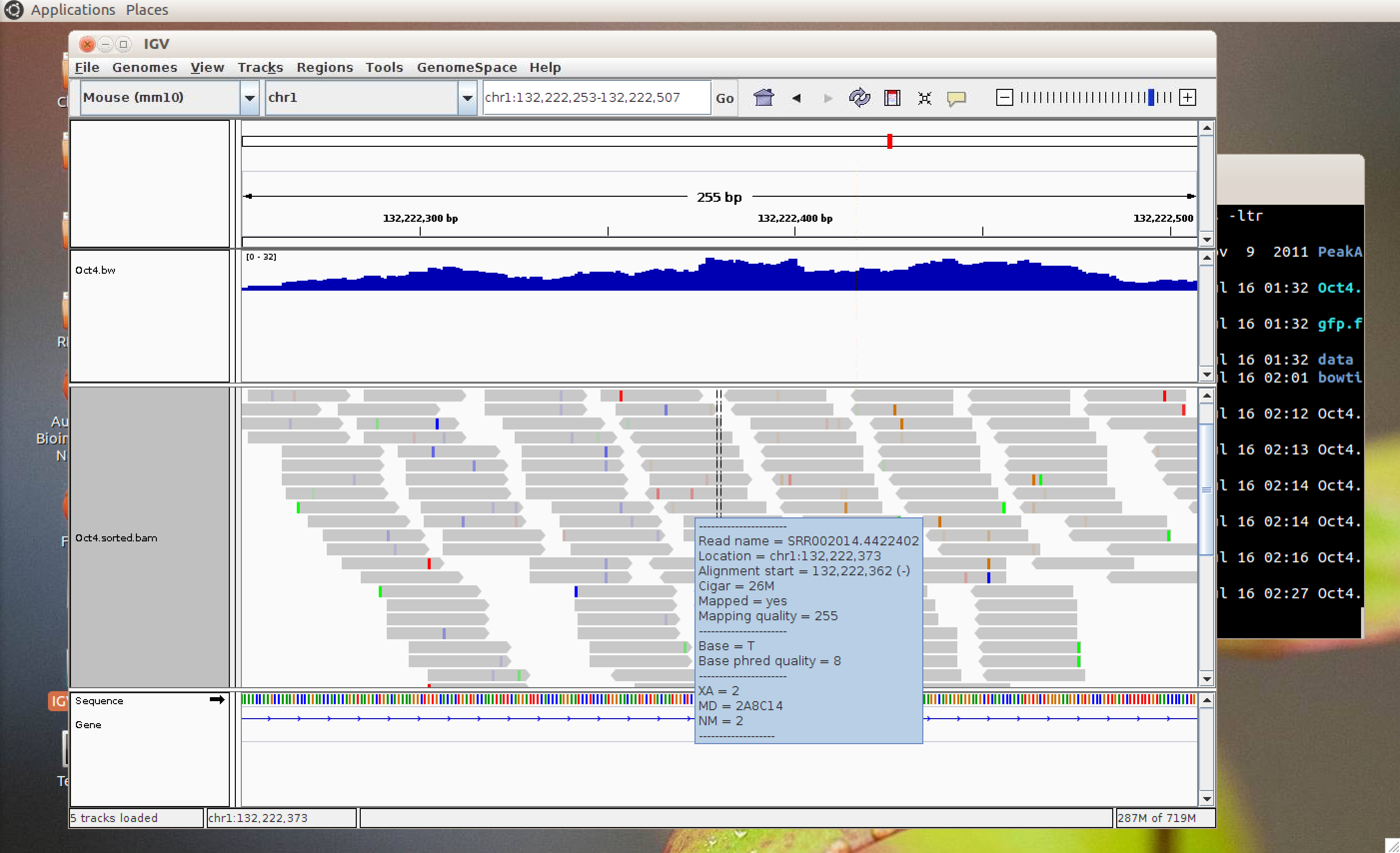
This allows for rapid visualisation of individual aligned reads across the entire genome. The inbuilt MACs peak caller is used for first pass peak calling. A selection of peaks is then validated in the lab by ChIP-qPCR experiments and peak-calling parameters can be adjusted based on these results. Overlap analysis with regions of interest can be performed in SeqMonk. Aligned sequence files are converted to BigWig format using customised Perl scripts and uploaded onto the NeCTAR Object Storage (Swift), which can then be loaded seamlessly on the UCSC Genome Browser for visualisation and further investigation. Once the sequence files are uploaded to the object storage, it can then be easily compared against public ENCODE datasets and UCSC genomic annotations to identify any potentially interesting correlations.
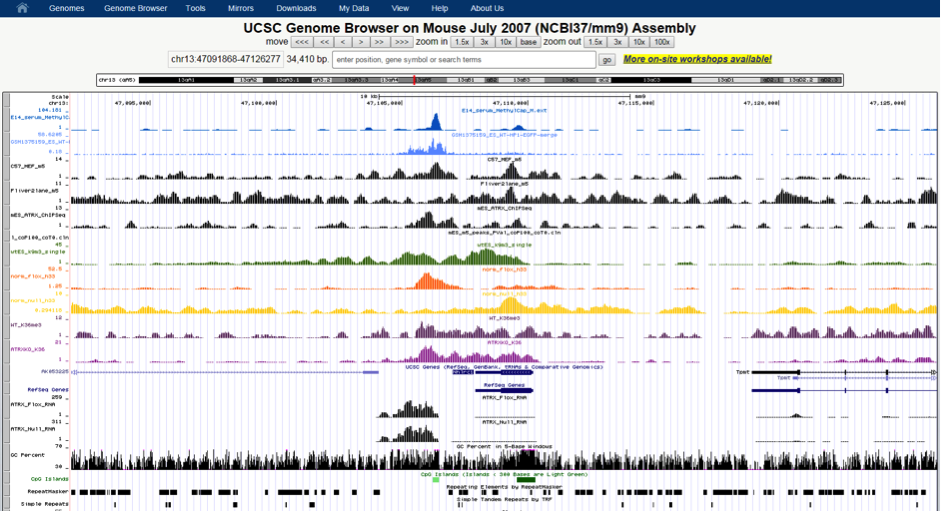
Aligned sequence visualisation using the UCSC Genome Browser.
The R@CMon team and the Monash Bioinformatics Platform supported the EpiC Lab by deploying a dedicated analysis instance on the NeCTAR Research Cloud based on the training environment first developed for the BPA-CSIRO Bioinformatics Training Platform. The open access and reusability of the training platform means it can be easily readapted to various analysis workflows. The R@CMon team and the Monash Bioinformatics Platform will continue to engage with the EpiC Lab as they grow and scale their analysis workflow on the NeCTAR Research Cloud.