Professor Jenkins’ research focuses on pancreatic cancer, an inflammation-associated cancer and the fourth most common cause of cancer death worldwide, with an extremely low 5% five-year survival rate. Typically studies look at gene expression patterns between normal pancreas and cancerous pancreas in order to identify unique signatures, which can be indicative of sensitivity or resistance to specific chemotherapeutic treatments.
“Using next generation gene sequencing, involving big instruments, big data and big computing – allows near-term disruptive change in the clinical treatment of pancreatic cancer.” Prof. Jenkins, Monash Health..
To date, gene expression studies have largely focused on samples taken from open surgical biopsy; a procedure known to be very invasive and only possible in 20% of pancreatic cancers. Prof Jenkins’ group, in collaboration with Dr Daniel Croagh from the Department of Upper Gastrointestinal and Hepatobiliary Surgery at Monash Medical Centre, recently trialled an alternative less invasive process available to nearly all pancreatic cancer patients known as endoscopic ultrasound-guided fine-needle aspirate (EUS-FNA) which uses a thin, hollow needle to collect the samples of cells from which genetic material can be extracted and analysed. The challenge then becomes to ensure gene sequencing from EUS-FNA samples is comparable to open surgical biopsy such that established analysis and treatment can be used.
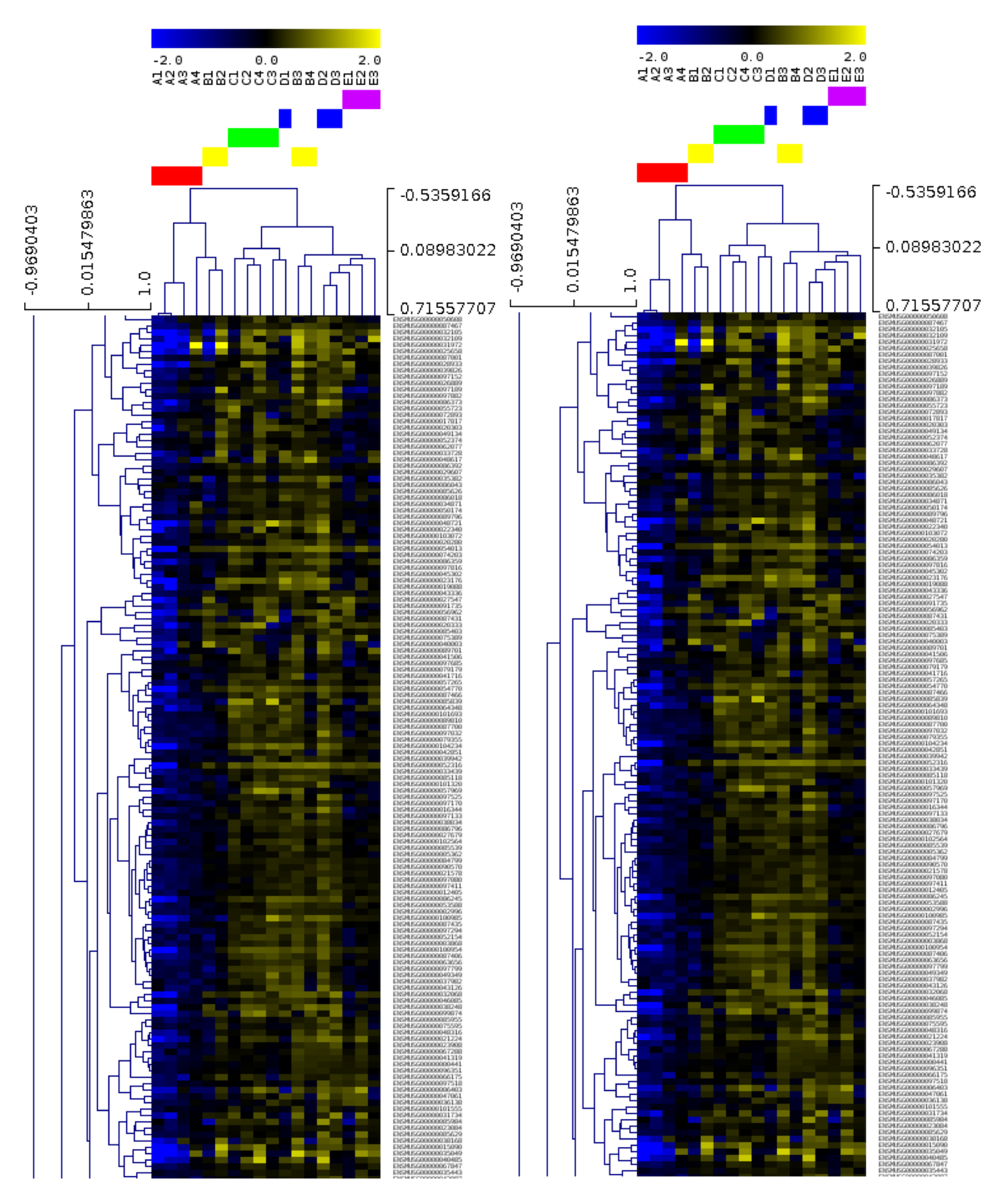
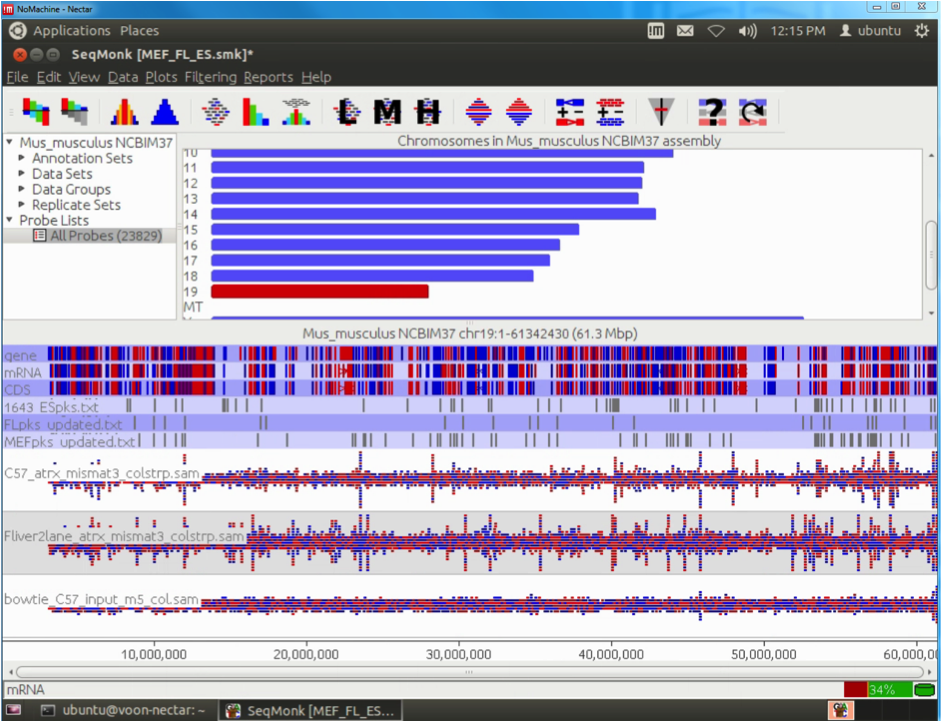
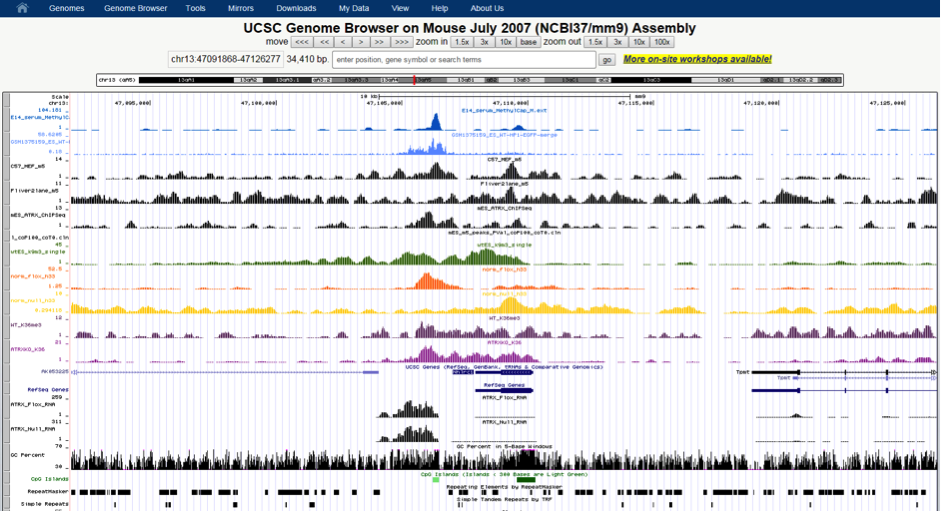
Twenty-four EUS-FNA-derived genetic samples from normal and cancerous pancreas were sequenced at the MHTP Medical Genomics Facility producing a total amount of 40Gb of raw data. Those data were securely transferred onto R@CMon by the Monash Bioinformatics Platform for processing, statistical analysis and computational exploration using state-of-the-art Bioinformatics methods.

Results thus far from this study show that data from EUS-FNA-derived samples were of high quality and also allowed the identification of gene expression signatures between normal and cancerous pancreas. Professor Jenkins’ group is now confident that EUS-FNA-derived material not only has the potential to capture nearly all of pancreatic cancer patients (compared to ~20% by surgery), but to also improve patient management and their treatment in the clinic.
“The current clinical genomics research space requires specialized high performance computational and storage infrastructure to support the processing and long term storage of those so-called “big data”. Thus R@CMon plays a major role in the discovery and development of new therapies and the improvement of Human health care in general.” Roxane Legaie, Senior Bioinformatician, Monash Bioinformatics Platform