At SuperComputing 2015 in Austin our network/fabric partner Mellanox announced R@CMon (Monash University) as a “HPC Centre of Excellence“. A core goal of the HPC CoE is to drive the technological innovations required for the next generation (exascale) supercomputing, whilst also ensuring that such an exascale computer is relevant to modern research. R@CMon is a stand out pioneer at converging cloud, HPC and data, all of which are key to the “next generation”.
“We see Monash as a leader in Cloud and HPC on the Cloud with Openstack, Ceph and Lustre on our Ethernet CloudX platform.” Sudarshan Ramachandran, Regional Sales Manager, Australia & New Zealand
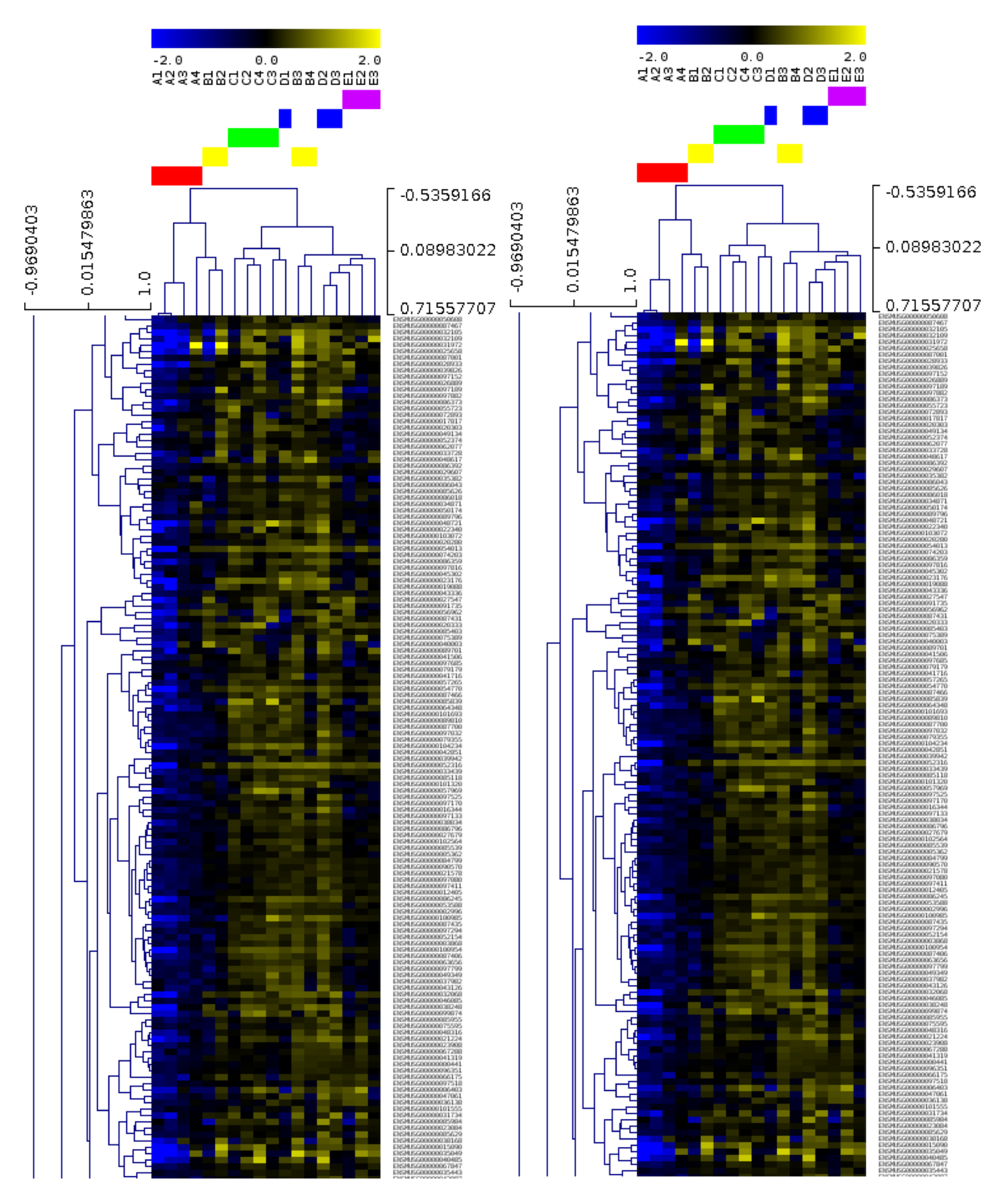
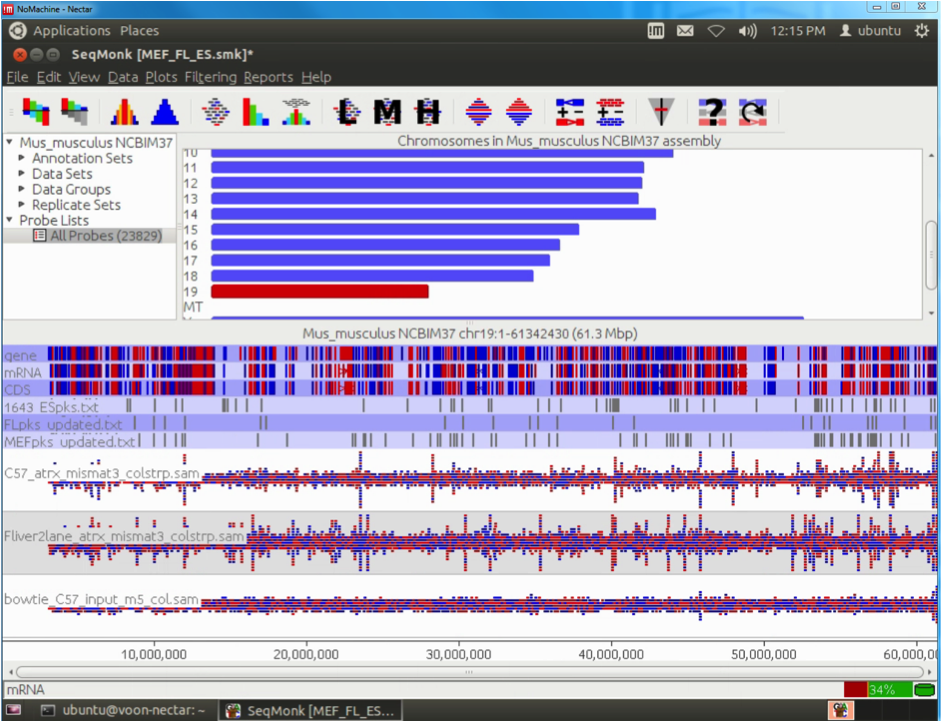
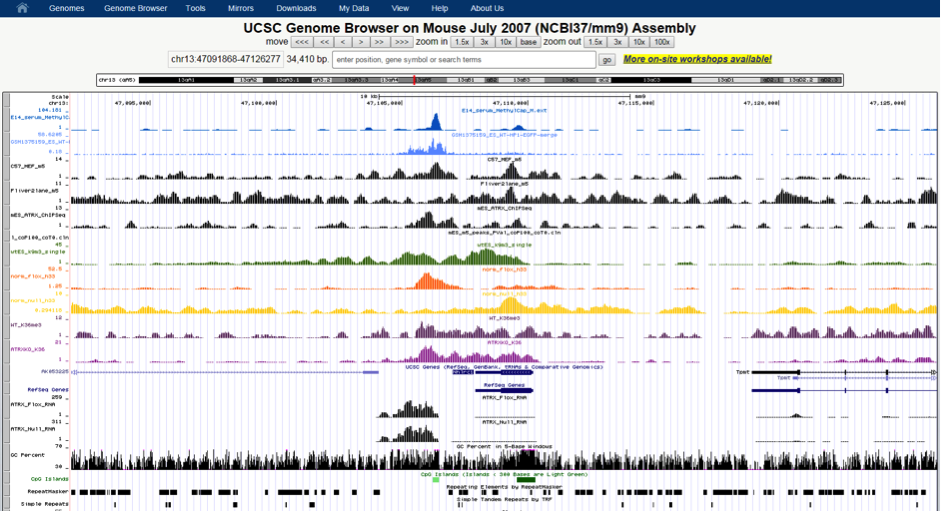
From a fabric innovation point of view, it has been a very productive and exciting 24months for R@CMon. By early 2014 the internal Monash University HPC system “MCC” was burst onto the Research Cloud, allowing a researcher’s own merit the be leveraged with institutional investment. It also represents a shift towards soft HPC, where the size of a HPC system changes regularly with time. Earlier this year we announced our early adoption of RoCE (RDMA over Converged Ethernet) using Mellanox technologies. The meant the same fabric used for cloud networking could also be used for HPC and data storage backplanes. In turn MCC on the R@CMon also enabled RDMA communications, that is, real HPC performance but on an otherwise orchestrated cloud.
Finally at the Tokyo OpenSack summit 2015, Mellanox announced R@CMon as debuting the World’s first 100G End-to-End Cloud. This technology eases scaling and heterogeneity of performance aspects. In particular, it sets the basis for processor and storage performance for peak and converged cloud/HPC needs. Watch this space!